Синдром Марфана – це спадкове захворювання, що передається по аутосомно-домінантним типом і характеризується ураженням сполучної тканини і її компонентів.
Хвороба Марфана викликається мутації гена, що кодує фібриліну -1.
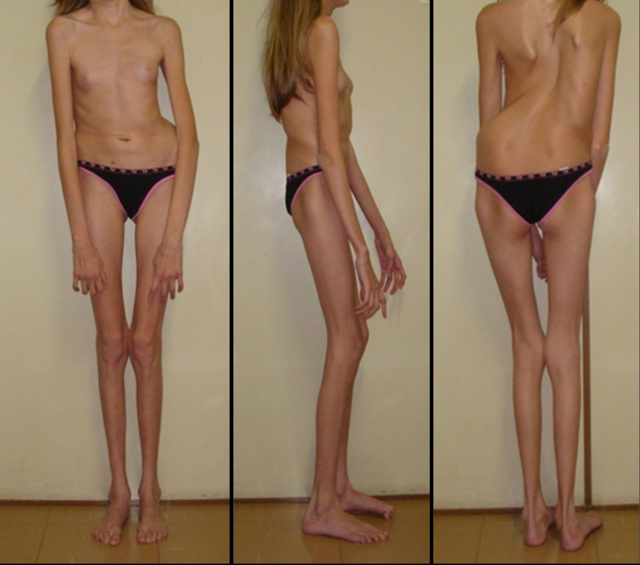
Люди з синдромом Марфана мають подовжені кінцівки, павукоподібні пальці і слабкий (недорозвинений) підшкірно-жировий шар і надгнучкі суглоби (див. Фото нижче).
Крім змін кістково-суглобової системи, характерні зміни зорового аналізатора і серцево-судинної системи. Також можливе ураження нервової, дихальної та інших систем.
Вперше описав цю патологію Вільямс, який помітив у своїх брата і сестри – випадання кришталика, при цьому вони були дуже високими і мали гіпермобільністю суглоби. Потім помітив Марфана, лікар – невролог, у якого протягом 20 років спостерігалася жінка з подібними симптомами, а потім ще 20 дітей.
Причини виникнення

Хвороба Марфана у дітей успадковується по аутосомно-домінантним типом (т. Е передається від батьків до дитини).
- Також можливі мутації за рахунок впливу на організм жінки факторів зовнішнього середовища (іонізуюче випромінювання, променева терапія, радіація).
- Причини виникнення і механізм розвитку захворювання недостатньо вивчені.
- Особлива роль відводиться порушення процесів обміну, в результаті яких накопичується в колагенових і еластичних волокнах велику кількість мукополісахаридів.
- Це веде до того, що сполучна тканина перерозтягується, легко піддається механічній дії і призводить до розвитку клінічної симптоматики.
Класифікація
Виділяють наступні форми хвороби Марфана:
Залежно від генної схильності:
- сімейна (патологія передається від батьків до дитини);
- спорадична (патологія викликана раптовим мутирование в геномі).
Залежно від проявів клініки:
- стерта, коли ознаки захворювання практично не виявляються і можуть бути не помічені протягом усього життя. Патологічні зміни виявляються в одній або двох системах.
- виражена, коли ознаки захворювання стосуються двох і більше органів і систем (серце, кістки і суглоби, легені, шкіра, очі).
Симптоми хвороби Марфана




На синдром Марфана у людей веде до їх виділенню в суспільстві своїм непропорційним будовою скелета. Для новонароджених на ранній стадії хвороби, характерні довгі пальці на руках, а до 7-9 років у дітей формується розгорнута клінічна картина.
У дорослих характерна різна симптоматика в залежності від системи поразки:
- Нервова система: біль у поперековій ділянці, головний біль, ураження симпатичної і парасимпатичної іннервації органів черевної порожнини і малого таза (слабкість кишкової стінки, нетримання сечового міхура у дитини). Також високий ризик розвитку інсульту, субарахноїдального крововиливу та розриву аневризм головного мозку.
- Сердечно – судинна система: пороки серця (звуження легеневої артерії, пролабирование стулок двостулкового клапана, дилатаційна кардіоміопатія, розширення меж серця (аорти і всіх її відділів), дефекти МЖП і МЖЖ перегородок. У хворих може розвиватися порушення ритму і провідності у вигляді аритмій.
- Опорно-руховий апарат: статура астенічної форми (діти худі), високий зріст у чоловіків 190 ± 10 см, у жінок 179 ± 8 см, слаборозвинений підшкірно-жировий шар, довгі пальці (павукоподібні), плоскостопість, череп і обличчя витягнуті і вузькі, недорозвиненість скул, порушення розвитку зубів і прикусу, витягнута нижня щелепа, готичне верхнє небо, гіпермобільність суглобів (дивіться на зображення вище). З віком дитини може прогресувати деформація хребетного стовпа, з розвитком сколіозу. Також може деформуватися грудна клітка, утворюється вдавлення – «груди шевця». Деформований тазостегновий суглоб нерідко призводить до інвалідності при ненадані своєчасному лікуванні.
- Орган зору: зміщення кришталика за рахунок слабкого зв’язкового апарату) на ранній стадії, сплощення рогівки, розвиток короткозорості або далекозорості, спазм акомодації, відшарування сітківки.
- Шкіра і м’які тканини: перерозтягнення шкіри з утворенням стрий атрофічного характеру. Вони виникають раптово, не пов’язані з коливання ваги людей, вагітністю і гормональним фоном. Шкіра липка, спітніла, з мармуровим відтінком. Підшкірно-жировий шар слабо розвинений, тому у хворих спостерігаються грижовоговипинання в області передньої черевної стінки.
- Дихальна система: розвиток бульозної емфіземи легенів, що виявляється кашлем, задишкою, розвитком дихальної недостатності і спонтанного пневмотораксу.
Інші ознаки:
діагностика
- Діагностика заснована на ретельному зборі анамнезу захворювання, вираженості клінічної картини, даних огляду, на результатах лабораторних та інструментальних методів досліджень.
- Збір анамнезу включає в себе: наявність в сім’ї даної патології (батьки, брати, сестри) або наявність факторів, що провокують мутацію в геномі людини.
- До лабораторних методів відносять: аналіз генотипу ДНК з мутує геном, визначення глікозаміногліканів в сечі.
- До інструментальних методів дослідження відносять:
- ЕКГ, служить для виявлення патології судин і серця (ССС). Виявляють характерні порушення ритму і провідності у вигляді миготливої аритмії, шлуночкової екстрасистолії, розвиток дилатаційною гіпертрофії міокарда лівого шлуночка.
- ЕхоКГ, також служить для виявлення патології ССС. Виявляють розширення аорти і її структур, пролабирование двостулкового клапана, збільшення розмірів лівої половини серця.
- УЗД серця проводиться для визначення ускладнень (расслаивающаяся аневризма).
- Рентген органів грудної клітини (зміни скелета, розширення порожнин серця, коренів легень тощо.)
- Комп’ютерна томографія, магнітно-резонансно-ядерна томографія дозволяє виявити патології кістково-суглобової, нервової системи, порушення кровообігу в судинах головного і спинного мозку.
Дані методи дослідження служать для виявлення критеріїв синдрому Марфана в різних органах і системах. Вони грають найважливішу роль для постановки і підтвердження діагнозу, а в подальшому, для визначення тактики лікування.
Існують наступні критерії діагностики синдрому Марфана:
| система | великі критерії | малі критерії |
| Опорно-руховий аппаратДолжни бути: 4 великих критерію, або 2 великих і 1 малий. |
|
|
| орган зору | зсув кришталика | Уплощенная рогівка, короткозорість, далекозорість, недорозвинення райдужки і війкового м’яза очей. |
| Серцево-судинна система | Розширення аорти і її структур | Пролабування двостулкового клапана, розширення клапана легеневої артерії в осіб, які не досягли 40 років, відкладення солей кальцію на стулках двостулкового клапана, розшарування аорти. |
| Дихальна система | відсутні | Раптово розвивається пневмоторакс (скупчення повітря в грудній клітці), верхівкові булли. |
| шкіра | відсутні | Повторне розвиток грижовоговипинань, атрофічні смуги. |
| Нервова система | Розширення судин оболонок спинного мозку в пояснічномкрестцовом відділі хребетного стовпа. | відсутні |
| генетичні зміни | Наявність даних критеріїв у батьків, дітей, братів, сестер, бабусь, дідусів. Наявність мутує гена, що кодує фібриліну 1. | відсутні |
Для постановки діагнозу «Синдром Марфана» враховується одна ознака з переліку великих критеріїв або малий критерій, характерний кожної з ураженої систем, крім опорно-рухового апарату, (необхідно, як мінімум, 4 критерії), а також наявність в сімейному анамнезі хворих з даною патологією .
Лікування синдрому Марфана
Від синдрому Марфана повністю позбутися і усунути механізм його розвитку неможливо. Лікування базується на поліпшенні загального стану хворого, усунення клінічних проявів і проведенні профілактичних заходів, що перешкоджають розвитку ускладнень.
Хворим з даними синдромам рекомендовано обмежити фізичне навантаження до низького рівня, або мінімального. Ризик появи патології серцево-судинної системи зростає при середніх і високих фізичних навантаженнях.
Слід обходити стороною і повсякденні навантаження, при яких можливе підвищення внутригрудного тиску, що веде до розвитку пневмотораксу (наприклад, підйом вантажів, підйом по поверхах).
Хворі з синдромом Марфана повинні консультуватися у різних фахівців, в залежності від клінічно уражених систем органів. Слід проходити медичні огляди кожні півроку протягом усього життя.
Медикаментозне лікування

- Медикаментозна терапія спрямована на усунення клінічної картини захворювання.
- З боку серцево-судинної системи рекомендовані β-адреноблокатори (наприклад: Анаприлин ), які знижують швидкість поширення пульсових хвиль при стрімко зростаючому розширенні аорти і зворотного потоку крові на двостулковому клапані або клапані аорти.
- β-адреноблокатори також надають позитивний ефект при порушенні ритму і провідності, в поєднанні з серцевими глікозидами.
- Але слід пам’ятати про існуючі протипоказання даних груп препаратів:
- Блокатори каналів кальцію використовуються при наявності протипоказань до В-адреноблокатори.
хірургія
Хірургічне лікування проводиться, якщо є ускладнення з боку серцево-судинної системи, з метою корекції уражених ділянок. Його проводять при Пролабування двостулкового клапана і розшаровуванні аорти.
При цьому здійснюється протезування двостулкового клапана.
У вагітних з тяжким перебігом хвороби Марфана, пологи дозволяються хірургічним шляхом.
профілактика
З профілактичною метою, щоб уникнути розвитку інфекційних ускладнень, утворення тромбів і тромбоемболії, призначаються антикоагулянти (гепарин), антибактеріальна терапія і вітамінотерапія.
- При синдромі Марфана з тяжким ураженням зорового аналізатора проводиться хірургічна корекція зору, після якої пацієнти повинні носити окуляри або контактні лінзи.
- Якщо виникають ускладнення, проводять лазерну корекцію глаукоми, катаракти, видаляють зміщується кришталик, замінюючи його на штучний.
- При функціональної дисфункції опорно-рухового апарату виникає необхідність стабілізування хребта за допомогою металевих пластин.
- При вираженій деформації грудної клітки проводиться торакопластіка.
- При протрузії тазостегнових суглобів виробляють внутрішнє протезування суглобів.
прогноз
Тривалість життя в середньому при синдромі Марфана становить 30-45 років.
Відомо, що це багато знаменитих особистості страждали даними синдромом. Це і Ганс Християн Андерсен – датський письменник, автор знаменитої Русалочки; Авраам Лінкольн – 16 президент США, Майкл Феллпс- відомий плавець, багаторазовий олімпійський чемпіон. А також відомі композитори – Нікколо Паганіні, Сергій Рахманінов.
Люди з даною патологією повинні ретельно стежити за своїм здоров’ям, постійно спостерігатися і консультуватися зі своїм лікуючим лікарем, уникати надмірних фізичних навантажень.
По мимо медикаментозного лікування, необхідно проведення профілактичних заходів з метою поліпшення загального самопочуття, підвищення імунітету, відповідний режим праці і відпочинку.
Відеозаписи по темі
цікаве

https://tvojajbolit.ru/kardiologiya/sindrom-bolezn-marfana-chto-eto-takoe-prichinyi-vozniknoveniya-simptomyi-i-lechenie-prognoz/
Синдром Марфана: причини, симптоми і лікування в статті гінеколога-ендокринолога Боровикова О. І
Дата публікації 9 квітня 2018 г.Обновлено 23 липня 2019 р
Синдром Марфана (Marfan; СМ) – генетично обумовлене захворювання, при якому відбувається системне ураження сполучної тканини. [1]

Етіологією захворювання є мутація в гені FBN1 (фибриллина 1), розташованому в короткому плечі п’ятнадцятої хромосоми в локусі 21.1. [2]
- Спадкування захворювання відбувається по аутосомно-домінантним типом, характеризується високою пенетрантностью (частотою появи гена) і різної експресивністю. [5]
- Співвідношення представників чоловічої статі і жіночої однакове.

При виявленні подібних симптомів проконсультуйтеся у лікаря. Не займайтеся самолікуванням – це небезпечно для вашого здоров’я!
Спостерігається постійно прогресуюче розвиток захворювання. У новонароджених дітей виявляються подовжені тонкі пальці на верхніх і нижніх кінцівках і подовжені тонкі кінцівки (доліхостеномелія). [1] У таких пацієнтів, крім доліхостеномелія, зазначається:
- підвищене фізичне розвиток;
- недолік ваги;
- подовжений череп;
- витягнуте лице;
- арахнодактилія (аномально подовжені вузькі пальці);
- слабкість і недорозвинення м’язової системи і жирової клітковини;
- незграбні рухи. [3]

Шкіра має підвищену еластичність, розхитані суглоби.
У більшості хворих спостерігається висока аркоподібне небо, зміни форми грудної клітини (воронкообразная, килевидная) і викривлення хребта (сколіоз в 60%, кіфоз (вигин хребта з утворенням горба), ювенільний остеохондроз), сплощення зводу стопи, аускультативні ознаки пороку серця (шуми) . [4] Довжина третього пальця руки – 10 см і більше (скринінговий тест у дітей 7-18 років): зростає співвідношення розмаху верхніх кінцівок до довжини тіла.

Офтальмологічні симптоми (короткозорість, підвивих кришталика в 75% випадків, його округлість або гіпоплазія, відшарування сітківки) і астенічні ознаки (втома, млявість) звертають на себе увагу з другого року життя, зміни форми грудної клітини з’являються у віці старше чотирьох років, патологія серця і судин виявляється в дошкільному віці. [1]
Майже у всіх хворих виявляються пороки серця і аорти. Часті стегнові та пахові грижі, ураження клапанів в венах, їх варикозне розширення, геморагічний синдром, рецидивуючі вивихи, ураження легеневої системи (мимовільний пневмоторакс, емфізематозних розширення легенів), опущення нирок. [2]

У чверті випадків зареєстровано зниження інтелекту, у половини пацієнтів виявляються порушення емоційно-вольової сфери. Часто з’являються депресивні стани, нейроциркуляторна дистонія. [3]
За даними багатьох досліджень, абсолютна більшість хворих з синдромом Марфана відзначають погіршення емоційного фону, втрату почуття радості і захопленості професійною діяльністю, часту зміну настрою, підвищену збудливість, відчуття тривоги. Результатом цього є зниження соціальної активності, погіршення якості життя і значне зменшення соціальної адаптації. [3]
У таких пацієнтів часто спостерігається трахеобронхиальная дискінезія (порушення дихальної системи) за рахунок слабкості сполучнотканинного каркаса бронхів.
Це проявляється рецидивуючими запальними захворюваннями бронхолегеневої системи, обструктивними порушеннями, бронхіальною астмою, емфіземою легенів (підвищений вміст повітря в легеневій тканині).
[4] Зустрічаються ускладнення, які проявляються скупченням повітря в грудній клітці, що супроводжуються здавленням легень і середостіння (серединної області грудної клітини), підшкірної емфіземою. Спостерігається неадекватна відповідь на бронхолітики. Обструктивні явища (непрохідність) зачіпають переважно верхні відділи респіраторного тракту. [3]
- Описано характерні зміни на електрокардіограмі, що включають синдром раннього збудження шлуночків, передчасні шлуночкові комплекси, нестабільність кінцевої частини шлуночкового комплексу в задненіжніх відведеннях. [3]
- Патологія ритму найчастіше проявляються блокадою правої ніжки пучка Гіса або змішаної екстрасистолією. [6]
- У хворих синдромом Марфана з патологією ритму серцевої діяльності і провідності синдром вегетативної дисфункції частіше протікає по ваготоніческому типу, у вигляді пресинкопальна, непритомних і астеновегетативних станів, хворобливих відчуттів в області серця, цефалгії напруги (головного болю) і часто поєднується з психопатологічними розладами. [4 ]
- Органи травлення також задіяні в патологічному процесі, що проявляється дискінезією (порушенням моторики) біліарного тракту зі зниженням моторики гладком’язових мускулатури, недостатністю кардії, грижовими випинаннями стравохідного отвору діафрагми, аномаліями жовчовивідних проток, доліхосігмой (збільшенням сигмовидної кишки), хронічний гастродуоденіт (запаленням слизової шлунка і дванадцятипалої кишки), дисбиозом (порушенням нормальної мікрофлори) кишечника, змінами підшлункової залози. [3]
- У пацієнтів з синдромом Марфана частіше, ніж у здорових людей, зустрічаються набуті аномалії нирок: підвищена рухливість нирок, нефроптоз (опущення нирки), піелоектазіі (аномальне розширення мисок), підвищена частота подвоєння нирок.
Більше половини ваги людини представлено сполучною тканиною, з неї складається наша головна опора – скелет, зовнішні покриви – шкіра. Судини, кров і лімфа теж складаються зі сполучної тканини.
До клітин сполучної тканини відносяться фібробласти і їх різновиди (остеобласти, хондроцити, одонтобласти, кератобласти), макрофаги (гістіоцити) і огрядні клітини (лаброцитів). [7]
Мезенхима – провідник конституційних, генетичних і епігенетичних складових життя людини. Патологія сполучної тканини детермінує певний патологічний дію на весь організм в цілому, на його фізіологію і його конституціональні особливості. [3]
При хвороби Марфана відбувається заміна нуклеотидів в гені, що містить інформацію про структуру пептиду фибриллина-1. Цей білок відноситься до гликопротеидам, бере участь в мікрофібрилярної комплексі, він забезпечує основу еластичних фібрил сполучної тканини.

- Міжклітинний матрикс дозволяє сполучної тканини підтримувати постійну структуру, в ньому знаходиться величезна кількість чинників зростання, які забезпечують постійне оновлення клітин.
- У великих судинах, зв’язковий апарат міститься велика кількість еластинових фібрил, ураження яких і дає основні клінічні прояви синдрому Марфана.
- При синдромі Марфана значно уражається трансформуючий фактор росту бета (TGF-β), порушується зв’язування його неактивної форми, що призводить до підвищення біоактивності даного чинника, з чим пов’язана поява багатьох проявів хвороби. [4]
- Патологія фибриллина призводить до патології формування волокон, що викликає втрату міцності і еластичності шкіри та інших сполучнотканинних структур.
- Зміна структури колагенових волокон призводить до порушення первинної ланки гемостазу у пацієнтів з синдромом Марфана. [6]
Є дані про дефекти мембранних і цитоплазматичних механізмів проведення сигналу безпосередньо в самому тромбоциті, що призводять до порушень агрегації (об’єднання). Показано наявність самостійного мембранного дефекту тромбоцитів, що протікає з порушенням реакцій вивільнення і транспорту внутрішньоклітинного кальцію. [6]
Еластичні фібрили мають цілком певні механізми участі в системі гемостазу. В судинах з низькою швидкістю зсуву відбувається адгезія ( «прилипання») тромбоцитів до еластину через фибронектин. [7] Реєструється зниження його рівня в крові у людей з синдромом Марфана.
Фібронектин, в свою чергу, утворюється в клітинах ендотелію і бере участь в наступних репаративних процесах, створюючи основу для виробництва інших компонентів сполучної тканини – фібробластів.
[4] Таким чином, абсолютно незаперечно участь судинної стінки в реакціях згортання крові, і неминучий висновок про можливі патологіях протікання нормальних гемостатических процесів при зміні стану її структурних компонентів і процесів судинної регуляції.
Відзначено роль гормонального дисбалансу в розвитку і посиленні дефектів сполучнотканинних структур. [3]
Тромботичні прояви детерміновані порушенням реології (в’язкості) крові в патологічно звивистих судинах брахиоцефальной зони. [3]
Ураження шлунково-кишкового тракту детерміновано тим, що ця система багата колагеном. Спостерігаються дискінезія біліарного тракту по гипомоторному типу, грижі стравохідного отвору діафрагми, аномалії жовчних шляхів, долихосигма, хронічний гастродуоденіт зі стертою клінічною картиною, схильністю до торпидной течією. [3]
- стерта (уражено не більше двох систем, зміни виражені незначно);
- виражена (незначні зміни в трьох системах або значне ураження однієї і більше систем).
- Виділяють різні типи за ступенем тяжкості:
- Частота важких форм – 1 до 25000-50000 (при загальній частоті діагностованих випадків 1 до 10000-15000).
- За характером перебігу:
- прогресуюча форма;
- стабільна форма.
Найчастіше перші ознаки синдрому Марфана проявляються ще в дитячому періоді, з віком відбувається прогресування симптомів, посилення клінічних проявів.
До найчастішим ускладнень синдрому Марфана відносяться:
- Зниження зору, аж до сліпоти, обумовлене слабкістю цинновой зв’язки (війкового паска) та підвивихи, вивихом кришталика. [7]

- Серцева недостатність по застійному типу, обумовлена порушенням скоротливості серцевого м’яза, недостатністю мітрального клапана. [6]
- Розриви великих судин, пов’язані з дилатацією (розширенням), витончення стінки судин. Найчастіше відбувається ураження аорти (в основному через зміни гемодинаміки при вагітності). [7]
- Аневризма аорти, що призводить до смерті хворих.

- Діагностика Синдрому Марфана грунтується на клінічних даних, виявленні змін в гені FBN1. [5]
- Часто при зборі генеалогічного анамнезу виявляються родинні випадки з прихованим перебігом захворювання. [1]
- Способи виявлення арахнодактіліі : [3]
- Симптом Steinberg (ознака першого пальця). Перший палець видно з-під hypothenar при напруженому кулаці.
- Симптом Walker-Murdoch (ознака зап’ястя). При обхвативании пензлем в області лучезапястного зчленування контралатеральної верхньої кінцівки перший палець заходить за п’ятий.
- Визначення п’ясткового індексу. Визначається за допомогою рентгенографії. Середня довжина п’ясті, поділена на усереднену ширину відрізка від другої до четвертої п’ясткової кістки. При нормальному співвідношенні цей показник відповідає 5,4-7,9, в той час, як при синдромі Марфана – більше 8,4.
У 2010 році група фахівців систематизувала міжнародні Гентського критерії для верифікації синдрому Марфана. Верифікація залежить від даних генеалогічного анамнезу. [3]
При відсутності генеалогічного анамнезу :
- збільшення діаметра аорти>, = 2 ϭ + ектопія кришталика = СМ;
- збільшення діаметра аорти>, = 2 ϭ + виявлені зміни в гені FBN1 = CM;
- збільшення діаметра аорти>, = 2 ϭ +>, = 7 системних ознак = СМ;
- ектопія кришталика + наявність змін в гені FBN1 + дилатація аорти = СМ;
При наявності генеалогічного анамнезу :
- Ектопія кришталика + випадок СМ в родині = СМ;
- >, = 7 системних проявів + випадок СМ в родині = СМ;
- збільшення діаметра аорти>, = 2 ϭ + випадок СМ в родині = СМ.
У п’ятнадцяти відсотках поява дитини з синдромом Марфана спорадичне (випадкове), у батьків можуть бути слабкі прояви. У родичів пацієнтів зустрічаються захворювання шлунково-кишкового тракту, ураження хребта, захворювання очей. [3]
При найменшій підозрі на синдром Марфана необхідна консультація офтальмолога.
В аналізі сечі таких пацієнтів відзначається підвищення рівня оксипроліну, глікозаміногліканів, але ці показники нізкоспеціфічни, можуть бути при різних дисплазиях сполучної тканини.
Виділення оксипролина є показником тяжкості захворювання. Спостерігається порушення згортання крові на тромбоцитарном рівні. [3]
Оцінка системних ознак залученості сполучної тканини
ПрізнакіБалли
| Спільне спостереження позитивних ознак Steinberg і Walker-Murdoch | 3 |
| Ознака Steinberg і Walker-Murdoch окремо один від одного | по 1 |
| Килевидное викривлення грудної клітини | 2 |
| Воронкоподобное викривлення, або асиметрія грудної клітини | по 1 |
| Медіальне зміщення медіальної кісточки, що приводить до уплощению стопи | 2 |
| сплощення стопи | 1 |
| Спонтанний пневмо- і гідроторакс (скупчення повітря і рідини в плевральній порожнині) | 2 |
| Розширення дурального мішка в крижовому і поперековому відділах | 2 |
| Підтверджена на рентгенограмах протрузія вертлюжної западини будь-якого ступеня | 2 |
| Зменшення відносини верхньої і нижньої частин тулуба (1,05 + викривлення хребта I-II ступеня. | 1 |
| Сколіоз або кіфосколіоз | 1 |
| Зменшення випрямлення в ліктьовому суглобі до 170 градусів і менше | 1 |
| Присутність трьох черепно-лицевих дізморфій з п’яти (доліхоцефаліческая форма черепа, запалі очі, антімонголоідний розріз очних щілин або зміщення очних щілин вниз, зменшення розмірів виличні кісток, ретрогнатия) | 1 |
| Розтяжки на шкірі | 1 |
| Міопічна патологія більше трьох дптр | 1 |
| Пролапс (прогинання стулок) мітрального клапана | 1 |
https://ProBolezny.ru/sindrom-marfana/
Хвороба Марфана: причини, симптоми і лікування
Хвороба (синдром) Марфана – це спадкова патологія сполучної тканини. Оскільки сполучна тканина є основою різних органів людини, то і прояви захворювання вельми різноманітні.
Найтиповішими є аномалії серцево-судинної системи, опорно-рухового апарату і очей. Діагностика синдрому Марфана вимагає використання самих різних методів. Лікування включає в себе медикаментозні і оперативні способи, причому спостерігатися хворий повинен у кількох лікарів.
З цієї статті Ви зможете дізнатися про причини, симптоми, діагностику і варіанти лікування даного захворювання.
Синдром Марфана формується ще у внутрішньоутробному періоді, дитина народжується вже з наявними порушеннями. Це, мабуть, найбільш часто зустрічається спадкова патологія сполучної тканини. Незалежно від статі і расової приналежності, частота виявлення хвороби Марфана коливається від 1: 5 000 до 1:10 000 населення (за різними даними).
причини
Основою захворювання є генетичний дефект: мутація гена, відповідального за синтез білка сполучної тканини фибриллина. Ген розташований на 15-й хромосомі.
Успадковується захворювання по аутосомно-домінантним типом. Аутосомное означає, що воно не пов’язане з підлогою, а домінантне – то, що мутація завжди проявляється. Ступінь прояви (поширеність порушень і їх вираженість) може варіюватися в різних межах, що пов’язано з генетичними особливостями.
Фібриліну надає сполучної тканини еластичність і розтяжність. Порушення його будови призводить до втрати міцності і пружності сполучної тканини, і вона перестає служити міцним каркасом. В першу чергу зміни торкаються стінок судин, зв’язкового апарату. Навіть невеликі фізіологічні навантаження стають позамежними для організму, сполучна тканина їх не витримує.
Всі випадки синдрому Марфана з точки зору причини поділяють на:
- сімейні: складають 75%, це передається з покоління в покоління мутація гена;
- випадкові (спорадичні): 25%, це вперше виникла мутація гена в роду, де раніше не було подібної патології.
симптоми
Клінічних проявів захворювання досить багато. Їх розглядають з точки зору ураження окремих органів і систем:
- опорно-рухового апарату;
- серцево-судинної системи;
- органу зору;
- нервової системи;
- бронхолегеневої системи;
- шкіри, м’яких тканин;
- інших органів.
Опорно-руховий апарат
 Для хворих з патологією сполучної тканини характерна гипермобильность суглобів.
Для хворих з патологією сполучної тканини характерна гипермобильность суглобів.
Хворі з синдромом Марфана зазвичай мають високий зріст, худорляві через недорозвинення підшкірно-жирової клітковини. Тулуб при цьому здається коротким, а кінцівки – непропорційно довгими. Розмах рук більше зростання на 5% і більше. Череп витягнутий (доліхоцефаліческая), пальці довгі павукоподібні (арахнодактилія). Особа витягнуте, вузьке, небо високе аркоподібне (готичне), вуха великі, нижня щелепа виступає вперед (неправильний прикус), зуби ростуть неправильно, очі глибоко посаджені.
Деформація скелета полягає в наявності лійкоподібної або килевидной грудної клітини, будь-яких змін в осі хребта (надлишковий лордоз, кіфози, кіфосколіози, сколіози), підвивихів хребців (особливо в шийному відділі), спондилолистеза (зміщення вищого хребця по відношенню до нижчого).
Характерно плоскостопість різних ступенів вираженості, розхитаність суглобів, надлишкові руху в них (наприклад, переразгибание), протрузія вертлюжної западини (поглиблення з глибоким зануренням головки стегнової кістки в області тазостегнового суглоба).
Зазвичай все це супроводжується зниженням м’язового тонусу.
Серцево-судинна система
Зміни в цій системі часто є найбільш важкими, які вимагають корекції в першу чергу, оскільки можуть бути загрозливими для життя.
Найчастіше синдром Марфана проявляється наявністю вад розвитку серця і великих судин (зокрема, аорти).
Дефекти будови серця полягають в порушенні перегородок серця і його клапанів (пролапс мітрального клапана, дефект міжшлуночкової або міжпередсердної перегородки, патологічне подовження хорд).
Порушення будови стінки аорти призводить до розвитку її дилятации (розширення) і розшарування стінок (аневризма аорти). З вроджених вад великих судин можуть спостерігатися коарктация аорти, стеноз (звуження) легеневої артерії.
Патологічний будова серця і судин супроводжується розвитком серцевої недостатності, порушенням серцевого ритму (надшлуночкова і шлуночкова тахікардії, фібриляція передсердь та інших), що вельми небезпечно для життя хворого. Приєднання інфекційних ускладнень загрожує розвитком інфекційного ендокардиту.
орган зору

Симптоми ураження очей дуже специфічні для даного захворювання. Поразка опорно-рухового апарату, серцево-судинної системи і очей складають типову тріаду симптомів при хворобі Марфана.
Хворі синдромом Марфана часто мають блакитні склери. Зір поганий зважаючи розвитку короткозорості високого ступеня, довжина очного яблука збільшена. Зіниці можуть бути не симетричними, тобто різними за розміром.
Однак цим поразки органу зору не обмежується. Розвиваються косоокість, вивих або підвивих кришталика, недорозвинення райдужки і війкових м’язів, сплощення рогівки. Можлива відшарування сітківки з розвитком сліпоти.
Нервова система
Поразка нервової системи в основному пов’язано з патологією будови стінки судин. Це призводить до порушення кровообігу, розвитку ішемічних або геморагічних інсультів (частіше – субарахноїдальних крововиливів).
Можливі часті непритомності. З аномалій можливі ектазія твердої мозкової оболонки, зокрема попереково-крижове менінгоцеле (випинання оболонок мозку під шкіру з утворенням кишені через дефект в хребцях).
Можливі відхилення в психомоторному розвитку, причому як в сторону затримки, так і в бік перевищення показників. Багато людей з синдромом Марфана мають високі показники інтелекту (вище, ніж середньостатистичний показник IQ в популяції).
Ще одним симптомом при хворобі Марфана можуть служити гіпофізарні розлади з підвищенням вмісту адреналіну в крові, розвитком акромегалії і нецукрового діабету.
бронхолегеневої системи
Поразка бронхів і легенів полягає в розвитку спонтанного пневмотораксу, дихальної недостатності, емфіземи легенів, апікальних булл (пухирчастих освіти в верхівках легенів). Всі ці зміни стають результатом порушення ангиоархитектоники легких через патологію сполучної тканини, підвищеною розтяжності і зниженою еластичності легеневої тканини.
Поразка шкіри і м’яких тканин
Часто хворі з синдромом Марфана мають атрофічні смуги на шкірі: хвилясті смуги на шкірі різної ширини і кольору (від білого до червоно-фіолетового). Мала кількість підшкірно жирової клітковини стає причиною малої ваги таких хворих.
Хворі з синдромом Марфана страждають рецидивними паховими і стегновими грижами.
інші органи
Патологія сполучної тканини стає причиною опущення нирок, сечового міхура і матки, варикозного розширення вен, призводить до утворення довгого і зі слабкою перистальтикою кишечника (що супроводжується запорами).
діагностика
 Лікар може запідозрити наявність синдрому Марфана у хворого за результатами огляду та обстеження.
Лікар може запідозрити наявність синдрому Марфана у хворого за результатами огляду та обстеження.
Діагностика синдрому Марфана вимагає обліку анамнестичних даних (в тому числі і спадкового анамнезу), даних огляду хворого, а також проведення різноманітних додаткових методів дослідження, що дозволяють виявити зміни в серці, судинах, головному мозку, легенів, очах. Для цього можуть знадобитися ЕКГ, ЕхоКГ (УЗД серця), рентгенографія грудної клітки і тазостегнових суглобів, КТ або МРТ головного та спинного мозку, серця і судин, офтальмоскопія, аортография і інші методи. Перелік досліджень залежить від наявності симптомів у конкретного хворого.
Існують діагностичні критерії цього захворювання: великі і малі. Певне поєднання великих і малих критеріїв і дозволяє підтвердити діагноз.
До великих критеріїв, наприклад, відносять підвивих кришталика, ектазія твердої мозкової оболонки, кілевідную або лійкоподібну грудну клітку, плоскостопість, протрузию вертлюжної западини і інші.
До малих критеріїв відносять надлишкову рухливість в суглобах, готичне небо, деформацію черепа, спонтанний пневмоторакс і інші.
Остаточний діагноз виставляється після застосування молекулярно-генетичного методу та встановлення мутації гена, відповідального за синтез фибриллина.
лікування
Лікування синдрому Марфана завжди вимагає одночасної участі декількох фахівців: кардіолога, кардіохірурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лікувальних процедур охоплює консервативні і оперативні способи лікування. Консервативні заходи спрямовані на профілактику ускладнень та підтримання нормального функціонування органів і систем, а оперативні припускають корекцію наявних анатомічних змін з метою запобігти виражене порушення функцій або навіть загрозу для життя хворого.
Хірургічні методи лікування:
- реконструктивні операції на аорті (при значному, більше 5 см, розширенні висхідної частини аорти і розшарування її стінки);
- протезування клапанів серця;
- видалення зміненого кришталика із заміною його на штучний;
- пластика хребта при вираженому сколіозі;
- ендопротезування кульшових суглобів;
- пластика грудної клітини (в останні роки заперечується її доцільність).
Хворому необхідний підбір окулярів або контактних лінз, іноді можлива лазерна корекція зору.
Медикаментозне лікування має патогенетичну і симптоматичну спрямованість.
Хворому з метаболічної метою призначають великі дози вітаміну С (1-3 г на добу), препарати з глюкозаміносульфатамі і хондроітінсульфата (Терафлекс, Структум, Хондроксид, Глюкозамин, Аміноартрін, Ельбона, Юніум), Карнітину хлорид 20% розчин, Бурштинову кислоту по 100 200 мг 2 рази на день, препарати магнію (наприклад, Магне В6), поливитаминно-мінеральні комплекси (з кальцієм, магнієм, цинком, міддю). Прийом цих речовин спрямований на нормалізацію обміну речовин, зміцнення сполучної тканини.

В цілому підбір методу лікування і асортимент вживаних лікарських засобів дуже індивідуальні. Все залежить від спектра клінічних симптомів і вираженості порушень у конкретного хворого.
Хворим з синдромом Марфана показана лікувальна фізкультура, але в строго дозованому кількості, щоб заняття приносили користь серцево-судинної та опорно-рухової систем, а не шкоду. Не можна займатися контактними і ігровими видами спорту (баскетбол, футбол), рекомендовано плавання.
прогноз
Захворювання невиліковно, але постійне динамічне спостереження за хворим, хірургічна корекція серцево-судинних, суглобових і офтальмологічних порушень дозволяють знизити ризик для життя, поліпшити її якість, дозволити займатися своєю професією. Деякі хворі не доживають до 40 років, що пов’язано з серцево-судинними ускладненнями, в інших випадках тривалість життя становить 70 років (особливо після проведення кардіохірургічних операцій).
Таким чином, синдром або хвороба Марфана – це спадкове захворювання, протягом якого багато в чому залежить від старанності медичного спостереження та своєчасності надання медичної допомоги. Зазвичай лікування вимагає залучення багатьох фахівців, але такий комплексний підхід дозволяє уникнути безлічі ускладнень і підвищити якість життя хворого.
https://doctor-neurologist.ru/bolezn-marfana-prichiny-simptomy-i-lechenie
синдром Марфана
 Синдром Марфана – генетично детерміноване захворювання, що характеризується ураженням або недорозвиненням сполучнотканинних волокон під час ембріогенезу і проявляється дисфункціональними змінами з боку зорового аналізатора, кістково-суглобової і кардіоваскулярної систем.
Синдром Марфана – генетично детерміноване захворювання, що характеризується ураженням або недорозвиненням сполучнотканинних волокон під час ембріогенезу і проявляється дисфункціональними змінами з боку зорового аналізатора, кістково-суглобової і кардіоваскулярної систем.
Синдром був відкритий в 1875 році офтальмологом з Америки Е. Вільямсом.
Він виявив повне зміщення кришталика ока від свого нормального положення у брата і сестри, які мали високий зріст і надмірну рухливість суглобів з народження.
Через кілька років дитячий лікар з Франції А. Марфана вів спостереження за дівчинкою 5 років з прогресуючими аномаліями скелета, надмірно довгими кінцівками і «павуковими пальцями».
Він дав чіткий опис патології, завдяки чому синдром отримав його ім’я.
Що це таке?
Синдром (хвороба) Марфана – аутосомно-домінантне захворювання з групи спадкових патологій сполучної тканини. Синдром викликаний мутацією гена, що кодує синтез глікопротеїну фибриллина-1, і є плейотропних. Захворювання характеризується різної пенетрантностью і експресивністю.
У класичних випадках особи з синдромом Марфана високі (доліхостеномелія), мають подовжені кінцівки, витягнуті пальці (арахнодактилія) і недорозвинення жирової клітковини.
Крім характерних змін в органах опорно-рухового апарату (подовжені трубчасті кістки скелета, гіпермобільність суглобів), спостерігається патологія в органах зору і серцево-судинної системи, що в класичних варіантах становить тріаду Марфана.
Без лікування тривалість життя осіб з синдромом Марфана часто обмежується 30-40 роками [2], і смерть настає внаслідок расслаивающейся аневризми аорти або застійної серцевої недостатності. У країнах з розвиненим охороною здоров’я хворі успішно лікуються і доживають до похилого віку.
Історія захворювання
У 1876 р симптоми невідомої патології були відзначені доктором Вільямсом, але клінічні спостереження проводилися набагато пізніше – в 1896 р педіатром з Франції А. Марфану. Лікар протягом 5-ти років оцінював стан дівчинки з невивченими раніше аномаліями, що полягають в прогресуванні дистрофії скелета і м’язової тканини.
До середини 20-го століття було безліч описаних випадків, коли у хворих спостерігалися симптоми, близькі до патології Марфана, і всі вони ставилися до захворювань спадкового типу.
Серед таких випадків – розшарування аорти, пороки серця, ектопія кришталиків, що супроводжуються деформацією кісток (грудної клітки, хребта) і зовнішніми відхиленнями від норми (високий зріст, худорлявість, довгі кінцівки).
Американським генетиком Маккьюсика було проведено детальне дослідження мутацій хромосом і відкрита нова група захворювань сполучної тканини.
Класифікація
Форми патології:
- стерта – у хворих є незначні зміни в 1 або 2 системах організму;
- виражена – наявність слабовираженних порушень в 3 системах або характерних патологічних розладів хоча б в 1-ій системі.
Характер перебігу синдрому:
- прогресуючий – з плином часу патологія наростає і посилюється,
- стабільний – ознаки хвороби протягом багаторічних спостережень залишаються незмінними.
Етіологічна класифікація:
- сімейна форма – успадковується по аутосомно-домінантним принципом;
- спорадична форма – синдром обумовлений випадковою мутацією генів під час зачаття.
Причини виникнення
Це аутосомно-домінантне стан, генетичне захворювання, передається від батьків до дитини через гени.
Викликаний мутаціями в гені FBN1. Мутації FBN1 пов’язані з широким континуумом фізичних функцій, від ізольованих особливостей до важкої і швидко прогресуючої форми у новонароджених.
Особливості розлади найчастіше виявляються в серці, кровоносних судинах, кістках, суглобах і очах. Деякі особливості – наприклад, розширення аорти (розширення основного кровоносної судини, яке переносить кров від серця до іншої частини тіла) – може бути небезпечним для життя. Також можуть бути порушені легкі, шкіра, нервова система. Патологія не впливає на інтелект.
Поширеність, частота народження становить 1 з 5 000 чоловік, включаючи чоловіків і жінок всіх рас і етнічних груп.
Близько 3 з 4 успадковують його, тобто отримують генетичну мутацію від батька, у якого вона є. Тип успадкування при якому хворий перший в родині-називається спонтанною мутацією.
Існує 50-відсотковий шанс, народження дитини з генетичної мутації від уражених батьків.
Люди з синдромом Марфана народжуються разом з ним, але особливості розладу не завжди присутні відразу. У деяких людей є багато особливостей при народженні, включаючи серйозні стану, такі як розширення аорти.
У інших менше симптомів, наприклад, молоді не мають ознак, поки не стануть дорослими. Деякі особливості, особливо ті, які впливають на серце і кровоносні судини, кістки або суглоби, з часом погіршуються.
Це робить дуже важливим отримання точної ранньої діагностики і лікування. Без цього людина наражається на ризик від потенційно небезпечних для життя ускладнень. Чим раніше почалося лікування, тим краще результати.
Майже половина людей, які страждають синдромом Марфана, цього не знають.
симптоми
Найхарактернішою ознакою синдрому Марфана є поєднання уражень опорно-рухової, зорової та серцево-судинної систем. Терміни їх появи варіабельні, а прояви різноманітні.
Зазвичай для постановки діагнозу буває досить присутності таких ознак:
- непропорційно довгі кінцівки;
- аневризма аорти;
- одно- або двостороння ектопія кришталика.
Однак крім цих ознак існує ще близько 30 інших проявів синдрому.

Поразки органів зору
Одно- або двостороння ектопія кришталика при синдромі Марфана виявляється у 80% пацієнтів. Зазвичай у таких людей розвивається короткозорість і астигматизм, але в деяких випадках виникає і далекозорість. Найчастіше порушення зору відбуваються на 4-му році життя дитини. Далі вони стійко прогресують.
Крім цих захворювань, при синдромі Марфана можуть виявлятися наступні патології органів зору:
- косоокість;
- гіпоплазія циліарного м’яза;
- колобома райдужної оболонки;
- збільшення розміру і сплощення рогівки;
- зміна діаметра судин сітківки;
- формування катаракти;
- виникнення глаукоми в молодому віці.
поразки скелета
У людей з синдромом Марфана можуть виявлятися наступні зміни в опорно-рухової системи:
- зростання набагато вище середнього;
- астенічний тип статури;
- довгий і вузький лицьовій скелет;
- довгі кінцівки і пальці;
- викривлення хребетного стовпа (туберкульоз, сколіоз, кіфоз та ін.);
- воронкообразная або килевидная деформація грудної клітки;
- маленький розмір щелепи;
- аркоподібними високе небо;
- надмірна гнучкість і рухливість суглобів;
- молоткообразная деформація пальців стоп;
- протрузія вертлюжної западини;
- плоскостопість;
- патології прикусу.
Середнє зростання людей з таким захворюванням при народженні може становити у дівчаток 52,5 см (в дорослому віці близько 175 см), у хлопчиків 53 см (в дорослому віці близько 191 см).
Через високий неба і малих розмірів щелепи у людей з синдромом Марфана можуть виникати порушення мови. Поразки скелета і суглобових структур призводять до появи артралгий і миалгий. Пізніше такі зміни підвищують ризик розвитку раннього остеоартриту.
Ураження серця і судин
Домінуючими і найбільш небезпечними проявами синдрому Марфана стають ознаки уражень серця і судин. З’являються через пошкодження структури стінок судин еластичного типу зміни (особливо аорти та легеневої артерії) і пороки розвитку клапанів, перегородок серця викликають такі симптоми:
- швидке настання втоми;
- прискорене серцебиття;
- стенокардичні болю з локалізацією в спині, верхньої кінцівки або плечі;
- холодні руки і ноги;
- аритмії;
- задишка.
При вислуховуванні тонів серця у таких пацієнтів можуть визначатися шуми, а при виконанні ЕКГ виявляються ознаки стенокардії. При синдромі Марфана може розвиватися кістозна медійна дегенерація мітрального або аортального клапана, що призводить до пролапсу цих клапанних структур.
Крім цього, у плода, який успадкував мутовані гени, з високою часткою ймовірності можуть розвиватися вроджені вади серця.
Іноді, при несприятливому перебігу такої неонатальної форми синдрому, у дитини виникає прогресуюча серцева недостатність, що призводить до летального результату до року життя.
Однак найбільш характерним для синдрому Марфана ураженням серцево-судинної системи зазвичай стає прогресуюче розширення, розшарування всходящей частини аорти і поява на ній аневризм.
Такі зміни провокуються ослабленням сполучної тканини, що призводить до кістозної дегенерації судинної стінки. Швидке прогресування таких поразок аорти може охоплювати всю її довжину і відгалужується від неї судини.
Нерідко таке ускладнений перебіг патології призводить до смертельного результату.
поразки легких
Хворі з синдромом Марфана не завжди мають проблеми з легенями. Однак в деяких випадках слабкість сполучної тканини альвеол приводить до їх подовження і перерастяжению.
Згодом у таких хворих може виникати спонтанний пневмоторакс, емфізема легенів і дихальна недостатність.
При відсутності своєчасної допомоги і лікування такі патології можуть ставати причиною настання летального результату.
Крім цього, у людей з синдромом Марфана може спостерігатися нічний апное, що супроводжується припиненням дихання уві сні на 10 секунд і більше.
Інтелектуальний розвиток і стан психіки
У більшості дітей рівень інтелекту відповідає нормі і IQ складає 85-115 одиниць. У деяких осіб з таким спадковим захворюванням рівень IQ істотно перевищує верхня межа норми.
Іноді у людей з синдромом Марфана можуть бути присутні ознаки нерівномірного інтелектуальної діяльності і деякі особливості особистості, що виражаються в завищену самооцінку, надмірної емоційності, плаксивості і дратівливості.
Ураження центральної нервової системи
Одним з наслідків синдрому Марфана може ставати дуральна ектазія, викликає розтягування і витяжкою сполучної тканини (оболонки), що огортає спинний мозок. Згодом ця патологія може призводити до появи болів і дискомфортних відчуттів в черевній порожнині або до слабкості й нерухомості нижніх кінцівок.
Поразки інших систем
Крім вищеописаних проявів синдрому Марфана в деяких випадках можуть виявлятися наступні зміни в інших органах і системах:
- атрофічні смуги на шкірі;
часто рецидивуючі стегнові та пахові грижі; - схильність до розтягування і розривів зв’язок, підвивихи і вивихів;
- аномальне розташування нирок;
- варикозне розширення судин;
- опущення матки і сечового міхура.
Більшість дітей з синдромом Марфана важко переносять фізичне навантаження і після неї нерідко відчувають болю в м’язах. М’язи у таких хворих можуть бути недорозвиненими. На тлі психоемоційного перенапруження у дітей можуть періодично виникати напади мігренеподібного головного болю. Крім цього, нерідко відчувається слабкість і ознаки гіпотонії.
діагностика
Виявленням синдрому Марфана займаються фахівці в області генетики, кардіології, офтальмології, неврології, ортопедії.
Діагностика патології включає збір анамнезу життя і хвороби, виявлення типових клінічних ознак, аналіз зовнішнього огляду і фізикальних даних, результатів кардіографічних і рентгенографічного обстеження, відвідування офтальмолога, складання родоводу у генетика.
Основні діагностичні методики:
- загальний аналіз крові та сечі – типові ознаки запалення;
- біохімічне дослідження крові дозволяє виявити дисфункцію певного органу, що виникла в результаті розвитку патологічного процесу, а також визначити першопричину захворювання і призначити правильне лікування;
- електрокардіографія відображає електричні потенціали, сформовані в працюючому серці;
- ехокардіографія – дослідження морфологічних і функціональних змін серця і його клапанного апарату;
- рентгенографическое й томографічне дослідження – інформативні діагностичні методики, які виявляють ураження кісток, суглобів, внутрішніх органів і м’яких тканин;
- аортография – рентгенологічного дослідження аорти з використанням контрастної речовини;
- УЗД внутрішніх органів,
- биомикроскопия і офтальмоскопія,
- молекулярно-генетичний аналіз.
Як лікувати синдром Марфана
Лікування синдрому Марфана завжди вимагає одночасної участі декількох фахівців: кардіолога, кардіохірурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лікувальних процедур охоплює консервативні і оперативні способи лікування. Консервативні заходи спрямовані на профілактику ускладнень та підтримання нормального функціонування органів і систем, а оперативні припускають корекцію наявних анатомічних змін з метою запобігти виражене порушення функцій або навіть загрозу для життя хворого.
Хірургічні методи лікування:
- реконструктивні операції на аорті (при значному, більше 5 см, розширенні висхідної частини аорти і розшарування її стінки);
- протезування клапанів серця;
- видалення зміненого кришталика із заміною його на штучний;
- пластика хребта при вираженому сколіозі;
- ендопротезування кульшових суглобів;
- пластика грудної клітини (в останні роки заперечується її доцільність).
Хворому необхідний підбір окулярів або контактних лінз, іноді можлива лазерна корекція зору.
Медикаментозне лікування має патогенетичну і симптоматичну спрямованість.
Хворому з метаболічної метою призначають великі дози вітаміну С (1-3 г на добу), препарати з глюкозаміносульфатамі і хондроітінсульфата (Терафлекс, Структум, Хондроксид, Глюкозамин, Аміноартрін, Ельбона, Юніум), Карнітину хлорид 20% розчин, Бурштинову кислоту по 100 200 мг 2 рази на день, препарати магнію (наприклад, Магне В6), поливитаминно-мінеральні комплекси (з кальцієм, магнієм, цинком, міддю). Прийом цих речовин спрямований на нормалізацію обміну речовин, зміцнення сполучної тканини.
Для лікування серцево-судинних порушень часто використовують β-адреноблокатори (пропранолол, обзидан, атенолол), блокатори кальцію (Ніфедипін, амлодипін, Лекоптин), інгібітори ангіотензинперетворюючого ферменту (Лізиноприл, Периндоприл, Еналаприл), антиаритмічні препарати (при порушенні ритму серця). При розвитку інфекційного ендокардиту показані антибіотики. Після оперативних втручань може знадобитися антикоагулянтна терапія для зниження згортання крові (фраксипарин, Клексан).
В цілому підбір методу лікування і асортимент вживаних лікарських засобів дуже індивідуальні. Все залежить від спектра клінічних симптомів і вираженості порушень у конкретного хворого.
Хворим з синдромом Марфана показана лікувальна фізкультура, але в строго дозованому кількості, щоб заняття приносили користь серцево-судинної та опорно-рухової систем, а не шкоду. Не можна займатися контактними і ігровими видами спорту (баскетбол, футбол), рекомендовано плавання.

прогноз
Прогноз життя хворих з синдромом Марфана визначається, в першу чергу, ступенем серцево-судинних змін, а також уражень скелета і очей.
Є високий ризик ускладненого перебігу, зниження тривалості життя (90-95% не доживають до 40-50 років) і раптової смерті.
Своєчасна кардіохірургічна корекція при синдромі Марфана дозволяє значно збільшити тривалість (до 60-70 років) і поліпшити якість життя хворих.
Хворі синдромом Марфана повинні перебувати під постійним наглядом лікаря і регулярно проходити діагностичне обстеження.
При синдромі Марфана показаний низький або середній рівень фізичної активності, що виключає заняття контактними видами спорту, спортивні змагання, ізометричні навантаження, підводне плавання.
Жінкам дітородного віку з синдромом Марфана необхідно пройти медико-генетичне консультування.
https://doctor-365.net/sindrom-marfana/